Removing the Hidden Data Dependency of DIA with Predicted Spectral Libraries
Abstract
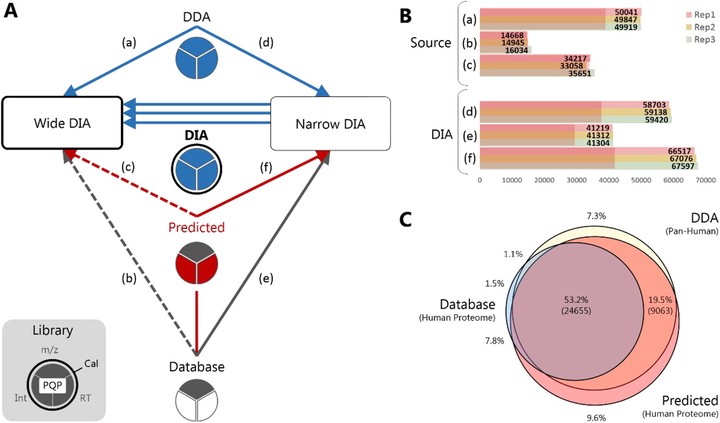
Abstract Data-independent acquisition (DIA) generates comprehensive yet complex mass spectrometric data, which imposes the use of data-dependent acquisition (DDA) libraries for deep peptide-centric detection. Here, it is shown that DIA can be redeemed from this dependency by combining predicted fragment intensities and retention times with narrow window DIA. This eliminates variation in library building and omits stochastic sampling, finally making the DIA workflow fully deterministic. Especially for clinical proteomics, this has the potential to facilitate inter-laboratory comparison.
Type
Publication
PROTEOMICS