Updated MS²PIP web server delivers fast and accurate MS² peak intensity prediction for multiple fragmentation methods, instruments and labeling techniques
Abstract
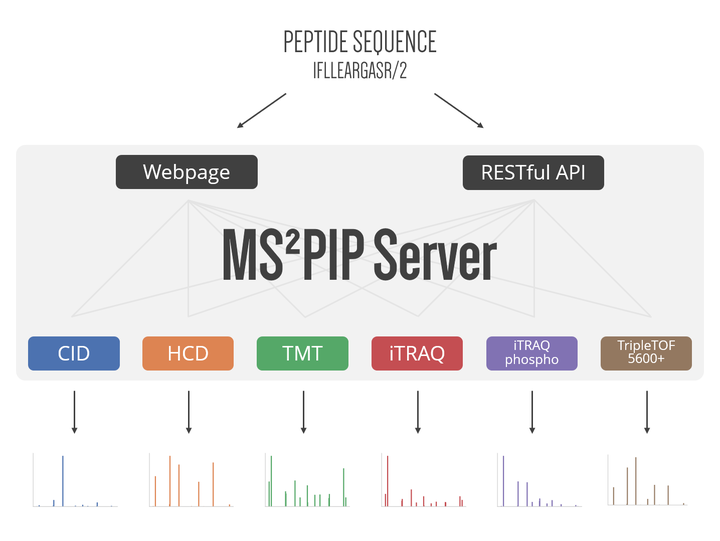
MS²PIP is a data-driven tool that accurately predicts peak intensities for a given peptide’s fragmentation mass spectrum. Since the release of the MS²PIP web server in 2015, we have brought significant updates to both the tool and the web server. In addition to the original models for CID and HCD fragmentation, we have added specialized models for the TripleTOF 5600+ mass spectrometer, for TMT-labeled peptides, for iTRAQ-labeled peptides, and for iTRAQ-labeled phosphopeptides. Because the fragmentation pattern is heavily altered in each of these cases, these additional models greatly improve the prediction accuracy for their corresponding data types. We have also substantially reduced the computational resources required to run MS²PIP, and have completely rebuilt the web server, which now allows predictions of up to 100 000 peptide sequences in a single request. The MS²PIP web server is freely available at https://iomics.ugent.be/ms2pip/.